Tutorial: Parametrizing the Trypsin/Benzamidine System
In this tutorial, we will be preparing sample SEEKR2 input files to determine the kinetics of binding between the receptor protein trypsin and the ligand molecule benzamidine. The parametrization will be done in the AMBER molecular mechanics forcefield.
Note
This tutorial is not a replacement for becoming proficient in using AmberTools. To learn how to use AmberTools, please visit the Amber Tutorials page at https://ambermd.org/tutorials/, review the Amber Manual at https://ambermd.org/Manuals.php, or both.
Identify your System
The first step for any SEEKR calculation is the identification of the target molecule (in this case, trypsin). If binding kinetics is the desired calculation (most common use of SEEKR), you must also identify at least one ligand molecule (in this case, benzamidine).
Trypsin binding to benzamidine is a very common model system to use for proposed computational algorithms that seek to estimate the kinetics of binding and/or unbinding between two molecules in a molecular system. The trypsin/benzamidine system is one of the first molecular systems that SEEKR programs were tested on (see [CIT2017]).
This tutorial is essentially a reproduction of system preparation in our 2017 paper.
Obtain the PDB structure
Once the target and ligand molecules have been defined, one must find Protein Data Bank (PDB) files of the molecular structures of the two molecules, ideally the structure of the two in the bound state. Such a structure does not, unfortunately, exist for the trypsin/benzamidine system in the PDB database.
Follow the link to the PDB page for trypsin/benzamidine: https://www.rcsb.org/structure/3PTB. We originally found this structure by searching for valid starting structures in scientific literature publications and by using the PDB’s own search engine.
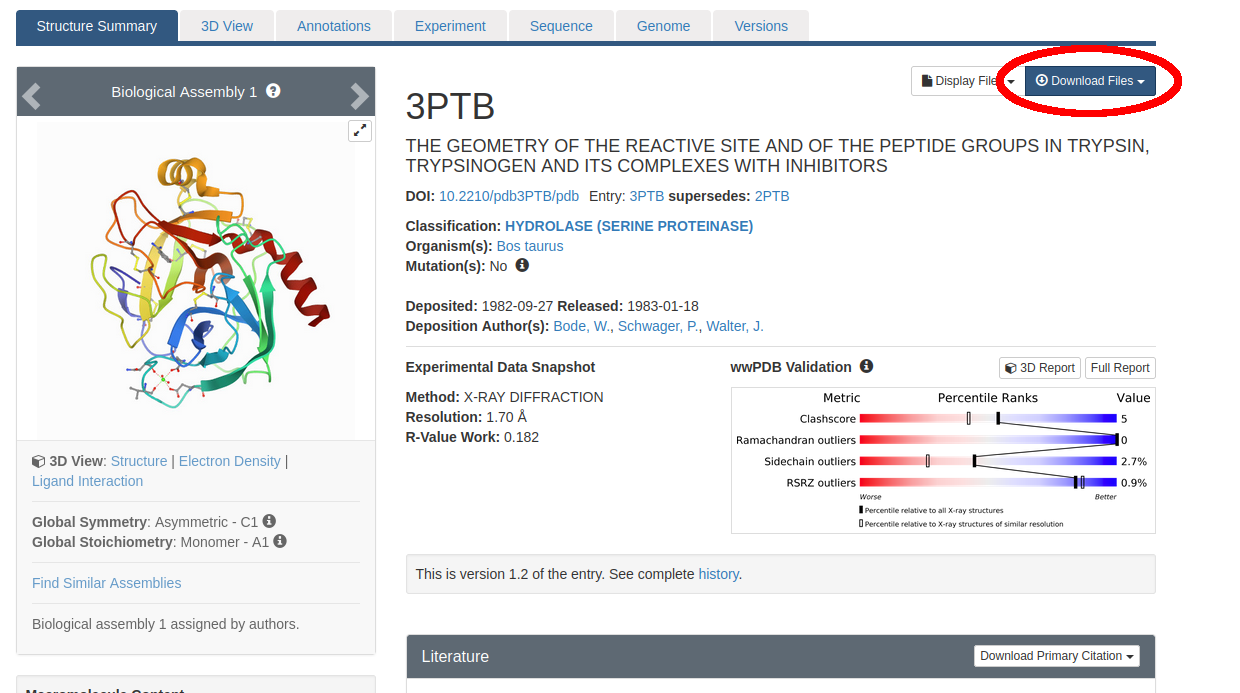
Figure 1: The Protein Data Bank entry we will use as a starting structure for our SEEKR2 calculations of Trypsin/Benzamidine (ID: 3PTB).
Click on “Download Files” near the top right (circled in red in Figure 1). Then select “PDB Format”. Save the file “3ptb.pdb” somewhere on your hard drive.
At this stage, many people will “clean” their PDB file using various tools. For instance, the Schrödinger commercial software, if available to you, includes a PDB cleaner. A free alternative is MolProbity (http://molprobity.manchester.ac.uk/).
Note
This process has been greatly simplified for demonstration purposes. When preparing your own system for a SEEKR2 calculation, many other procedures should be observed, including, for instance, assigning the correct protonation states to histidine residues. You should either consult an expert at preparing molecular dynamics (MD) simulations, or become an expert yourself before proceeding with your own system(s) of interest.
Since LEAP will not be able to handle the structure 3ptb.pdb as-is, and that structure doesn’t represent the bound state, we’ve provided a processed PDB file that you can download and proceed with:
Download 3ptb_processed.pdb.
This PDB has benzamidine in the bound state, which was obtained from one of our earlier SEEKR papers (see [CIT2017]).
Note
What should you do if your PDB does not have the ligand in the bound state? The automated software to handle this situation is under active development by the SEEKR team. However, at this time, you will need to use the so-called “ratchet method” to bring the ligand into the bound state using SEEKR2 itself. There is no tutorial for how to perform the ratchet method, though with enough interest, the SEEKR team may be persuaded to move it higher on the priority list.
Use Antechamber to Parametrize Ligand
SEEKR is often run on systems that contain at least one small molecule, which must be parametrized. One way to do this is with the Antechamber program in AmberTools.
Here, we will use Antechamber to parametrize our ligand: the small molecule benzamidine.

Figure 2: Benzamidine
Antechamber requires a structure file to parametrize the small molecule. In this case, we will use a PDB file of just the benzamidine.
Download ben.pdb.
Save ben.pdb somewhere on your hard drive. This molecule contains optimized molecular geometries.
Next, see if you have Antechamber:
which antechamber
If this returns a path, you should be good. Otherwise, you’ll need to install AmberTools. If antechamber exists on your computer, run the following command:
antechamber -i ben.pdb -fi pdb -bk BEN -o benz.mol2 -fo mol2 -c bcc -nc 1 -at gaff2
Each argument means the following information:
-i ben.pdb - Take the ben.pdb file as input.
-fi pdb - The format of ben.pdb is in PDB format.
-bk BEN - The component/block ID for benzamidine in the PDB file is “BEN”.
-o benz.mol2 - Specify output file name of benzamidine molecule.
-fo mol2 - Output the benz.mol2 file in MOL2 format.
-c bcc - Use the AM1-BCC semi-empirical method to assign partial charges of the atoms.
-nc 1 - This molecule has a net molecular charge of +1 due to its protonation state in aqueous environments at pH 7.
-at gaff2 - Use the GAFF2 small-molecule force field
Note
Semi-empirical methods for assigning charges are quick and easy, but are probably one of the least accurate methods for assigning partial charges to a molecule. For your own molecules, consider looking into more accurate levels of quantum calculations to obtain partial charges such as Hartree Fock with Density Function Theory (HF-DFT) or Møller-Plesset 2 (MP2). These types of calculations involving “higher” levels of quantum theory must be done with quantum calculation software such as Gaussian or GAMESS. Incorporation of parameters from quantum calculation software is a subject beyond the scope of this tutorial.
Use the parmchk2 program to generate a frcmod file, which LEAP will need to create the bound system:
parmchk2 -i benz.mol2 -f mol2 -o benz.frcmod
Then, generate a “.lib” file which will contain a library of forcefield parameters for the benzamidine molecule. Enter the following commands into the terminal:
tleap
source leaprc.gaff2
BEN = loadmol2 benz.mol2
saveoff BEN benz.lib
quit
Use LEAP to Create the Forcefield Parameters for the Solvated System
If you haven’t yet downloaded
3ptb_processed.pdb, please do so now.
Note
You should remove all CONECT records from your PDB files before using LEAP.
Now with a text editor, copy the following script to a file named leaprc:
source leaprc.protein.ff14SB
source leaprc.gaff2
source leaprc.water.tip4pew
set default PBRadii mbondi2
loadoff benz.lib
loadamberparams benz.frcmod
WAT = T4E
HOH = T4E
loadAmberParams frcmod.ionsjc_tip4pew
loadAmberParams frcmod.tip4pew
mol = loadpdb 3ptb_processed.pdb
solvateoct mol TIP4PEWBOX 8
addIons2 mol Cl- 0
saveamberparm mol tryp_ben.prmtop tryp_ben.inpcrd
savepdb mol tryp_ben.pdb
check mol
charge mol
quit
Then, run LEAP with the following command:
tleap -f leaprc
Let us consider a few key lines within this LEAP script:
source leaprc.protein.ff14SB - We are using the Amber14 protein parameters, which is one of the most recently generated parameter sets, and seems to perform quite well in SEEKR.
source leaprc.water.tip4pew - We are using the TIP4Pew water model, which, in theory, should be more accurate than the TIP3P water model.
set default PBRadii mbondi2 - The Born radii must be set in order to generate PQR files at a later step.
solvateoct mol TIP4PEWBOX 8 - We choose to use a solvated octahedron instead of a solvated box. This is because truncated octahedra are a more efficient use of space - less water molecules will be required to isolate the solute molecules from their periodic images. In fact, using an octahedron instead of a box usually cuts the number of water molecules in half, dramatically speeding up the simulations. We also create a buffer of 8 Angstroms around the protein. This number should be sufficiently large, but you could increase to 10 or even 12 if you want to be extra safe.
addIons2 mol Cl- 0 - Since this system is positively charged, this command will add chloride counterions until the system is neutrally charged. When consulting the publication that measured the kinetics of trypsin/benzamidine binding experimentally, we found that the system should contain a low concentration of calcium chloride and other buffers. Due to the low concentrations and difficulty of dealing with divalent ions in solution, we elected to merely add these counterions, with no additional ions mentioned in the publication. However, in general, it is a good idea to carefully consider your ions to recreate experimental or physiological conditions closely, if possible.
If everything runs correctly, the files tryp_ben.prmtop and tryp_ben.inpcrd should be generated.
Note
The tryp_ben.prmtop and tryp_ben.inpcrd files are essentially equivalent to those which exist in the SEEKR2 github repository at https://github.com/seekrcentral/seekr2.git located in seekr2/seekr2/data/trypsin_benzamidine_files/mmvt. However, since better practices were used in the preparation of the parameter/topology and coordinate files in the git repository, you should use those files for any production calculations, NOT the files generated in this tutorial.
Note
At this point, for your own systems, you should consider using the QMrebind tool (https://github.com/seekrcentral/qmrebind.git) to improve the forcefield parameters of the ligand within the binding site. A tutorial for QMrebind can be found here: https://qmrebind.readthedocs.io/en/latest/index.html. You may also read the QMrebind publication, and see its successful application to interesting systems here: https://chemrxiv.org/engage/chemrxiv/article-details/64d2d5bd69bfb8925a9adabf
Create the PQR files used in Brownian Dynamics
The final step, once you have parametrized your system, is to create the PQR files which will be used by the Brownian dynamics software to compute the binding rate constant (k-on).
AmberTools has a program for this purpose named ambpdb. Run the following command:
ambpdb -p tryp_ben.prmtop -c tryp_ben.inpcrd -pqr > tryp_ben_all.pqr
You can run ambpdb -h for an explanation of these arguments.
This will create a PQR file, which is almost identical to a PDB file, although instead of having a beta and occupancy column, the last two columns of the file represent the charge and radius of each atom.
Open the file tryp_ben_all.pqr in a text editor (like vim, emacs, or gedit).
Scroll way down until you find the BEN residue name (or perform a search in the document)
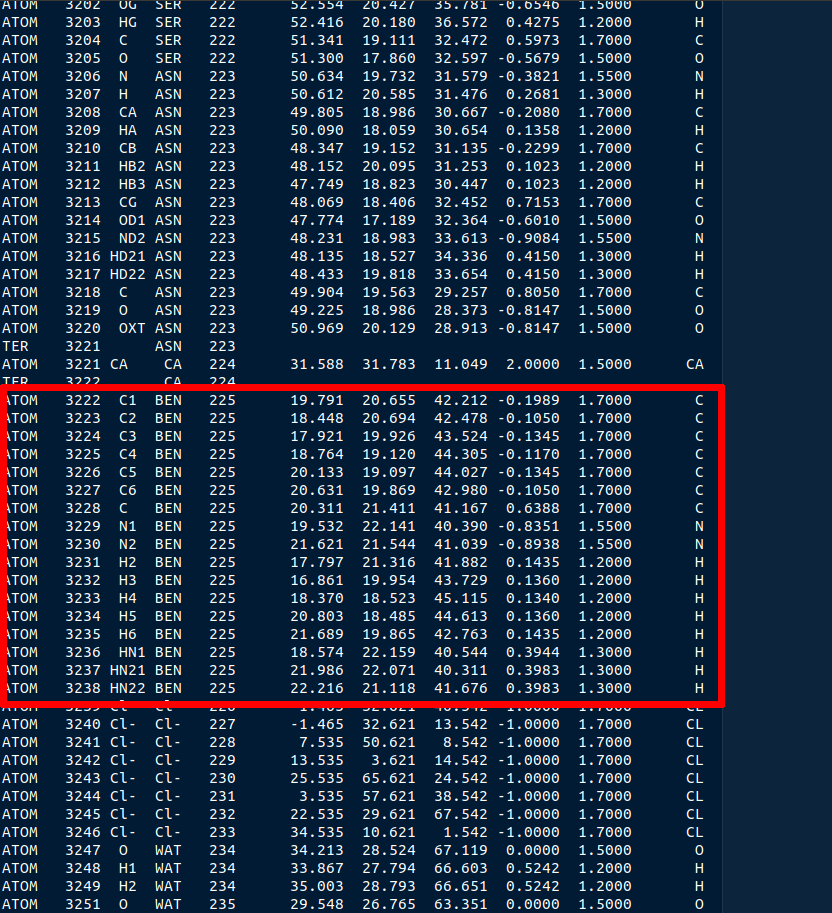
Figure 4: The BEN residue in the tryp_ben_all.pqr file.
Select all atoms with the BEN residue name and copy them over into a new file. Name that file tryp_ben_ligand_one_resid.pqr. The reason for this name will become clear in a moment.
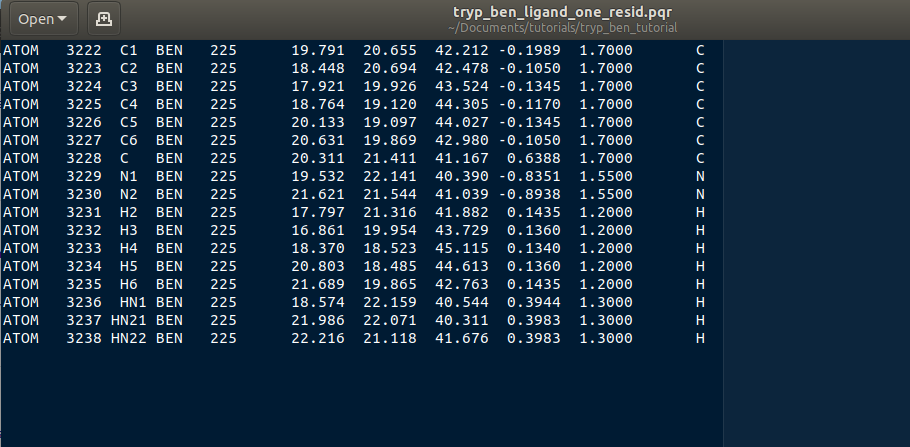
Figure 5: The BEN residue in the tryp_ben_pqr_one_resid.pqr file. Notice that the atom index and residue numbering are incorrect. We will correct these momentarily.
Next, back in tryp_ben_all.pqr, select the entire protein (all atoms above the BEN residue, including the calcium ion, since it is bound to the protein) and copy them over to a new file. Name that file tryp_ben_receptor.pqr. This new file will not have the BEN residue nor any water molecules in it.
Note
You should delete all lines in the PQRs that don’t begin with ATOM or HETATM. Browndye will have trouble with lines that begin with other words, such as TER or CRYST.
Now back to the file named tryp_ben_ligand_one_resid.pqr. Notice that this file has incorrect atom and residue numbering. More importantly, there is a feature of Browndye (the Brownian dynamics software that SEEKR2 uses) which lumps all charges of a residue into the same point (test charge). This may be fine for a protein such as in the file tryp_ben_receptor.pqr, but for a small molecule like benzamidine, we can improve accuracy by considering the point charges on each atom. This is accomplished by numbering each atom with a different residue number.
A script performs this automatically within Seekrtools, named pqr_resid_for_each_atom.py:
python /PATH/TO/seekrtools/seekrtools/scripts/pqr_resid_for_each_atom.py \
tryp_ben_ligand_one_resid.pqr tryp_ben_ligand.pqr
Obviously, change “/PATH/TO/seekrtools” to the actual path to your own instance of the Seekrtools git repository.
Now, open the file named tryp_ben_ligand.pqr. You will see that it is numbered correctly, with each atom assigned its own residue number.
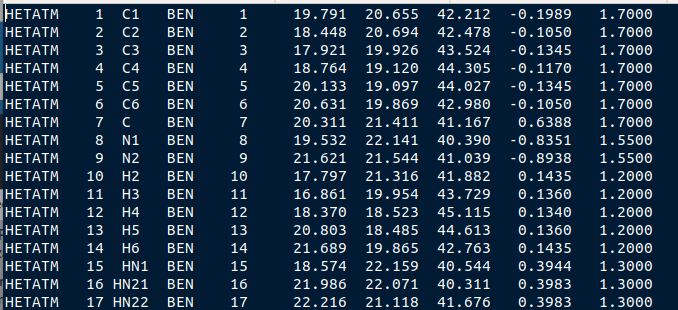
Figure 6: The atoms within this benzamidine PQR file are numbered correctly. The different residue numbers for each atom enhances accuracy in the Brownian dynamics calculations.
Now, the system is parametrized and ready for equilibration. You may proceed to the next tutorial.
Download any Missing Files
If anything went wrong with any steps above, you can download the files below to use for later tutorials.
Votapka LW, Jagger BR, Heyneman AL, Amaro RE. SEEKR: Simulation Enabled Estimation of Kinetic Rates, A Computational Tool to Estimate Molecular Kinetics and Its Application to Trypsin-Benzamidine Binding. J Phys Chem B. 2017 Apr 20;121(15):3597-3606. doi: 10.1021/acs.jpcb.6b09388. Epub 2017 Mar 3. PMID: 28191969; PMCID: PMC5562489.